Overview of Sarcoidosis: Special Attention to Musculoskeletal
Monsoon Rashid
Sarcoidosis is a multisystem disorder that can affect any organ of the body with the predilection to lung and intrathoracic lymph node. The hallmark of sarcoidosis is the presence of noncaseating granuloma, a cluster of macrophages, epithelioid cells, mononuclear cells, and CD4+ T cells with a few CD8+ T cells in the peripheral zone.1 Rheumatic manifestations of sarcoidosis may not be common, but rheumatologists deal with a considerable number of patients with sarcoidosis as a new case—these patients are referred for evaluation of joint pain or treatment of sarcoidosis when they need immunosuppressive therapy. This review article will discuss mostly rheumatic manifestations and treatment of sarcoidosis.
Overview of Sarcoidosis: Special Attention to Musculoskeletal
Monsoon Rashid
History
The word sarcoidosis comes from the Greek word “sarc,” meaning “flesh,” and “oid,” meaning “like”— in other words, “flesh-like.” It took many years to establish the name of sarcoidosis. Initially different cases of sarcoidosis were published by different investigators in different names. The first case was published by Jonathan Hutchison om 1875, where a 58-year-old coal-wharf worker visited Jonathan Hutchinson for skin diseases complaining of purple skin plaques—these plaques had gradually developed over the preceding two years somewhat symmetrically on his legs and hands. Hutchinson published his important case in 1898 as "Archives of Surgery” (Hutchinson). 'Hutchinson referred to this as “Mortimer's Malady” (lupus vulgaris multiplex non ulcerans et non-serpiginous). Another example of this was a 64-year-old woman who had multiple, raised, dusty-red patches which have no tendency to inflame or ulcerate. Hutchinson thought about tuberculosis or lupus as a differential, but noticed the disease was very different.
In I889, the Frenchman Ernest Besnier described a skin condition that he called lupus pernio. In 1897, Caesar Boeck presented a similar type of skin lesion and named the disease skin sarcoid; Boeck was the first person to do a biopsy on the lesions. The biopsy showed sharply defined foci of “epithelioid cells with large pale nuclei and also a few giant cells.”2 Later, many published case reports supported that sarcoidosis was a multisystem disorder rather than simply a skin condition.
Etiology
The exact cause of sarcoidosis remains unclear. As sarcoidosis mostly involves lung, skin, and eyes airborne antigens play a significant role. Studies show that for the development of sarcoidosis, there is an association with wood burning stoves, tree pollen, insecticides, metalworking, handling of building supplies. Investigators found the antibodies of many microorganisms like mycobacterium and propionibacterium in the serum of the patients—this supports the relation of infectious etiologies with the development of sarcoidosis. Case control etiologic sarcoidosis study (ACCESS) found that patients who had siblings or parents with sarcoidosis developed the disease five times more than the control group. Multiple studies showed an association between sarcoidosis and specific gene products including Class I HLA-B8 Ags, Class II HLA Ags, butyrophilin-like 2 (BTNL2) gene.3
Immunopathology
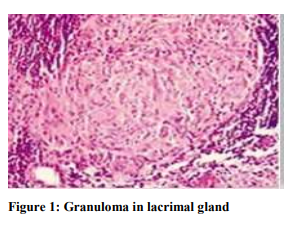
The fundamental abnormality in sarcoidosis is the development and accumulation of granuloma. Well-developed sarcoid granuloma consists of tightly formed epithelioid- and multinucleated-giant cells (MGCs) encircled by lymphocytes, especially CD4+ T helper (Th) cells, but also rare CD8+ T cells and B cells (Figure 1). Most sarcoid granulomas gradually resolve and leave few or no residual manifestations of previous inflammation. Small amounts of central fibrinoid necrosis may be seen. Large amounts of necrosis suggest an alternate diagnosis or necrotizing sarcoid granulomatosis.3
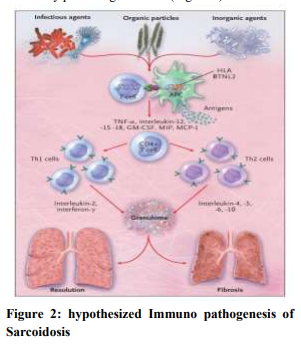
When genetically susceptible persons are exposed to a specific environments CD4+ cells interact with antigen presenting cell and initiate the process of formation of granuloma. CD4+ cells activate T helper cell 1 and T helper cell 2 which secret cytokines and chemokines most importantly interleukin-2 and interferon-γ, Tumor necrosis factor (TNF)-α. These products augment the local cellular response and ultimately produce granuloma (Figure 2).3
Epidemiology
Analysis of Optum healthcare database which included the patient with sarcoidosis above 18years of age showed among 29,372 adult patients 14,700 (55%) were above 55yrs of age at the time of diagnosis. The incidence and prevalence rates were higher for African Americans (17.8 and 141.4 per 100,000, respectively) than for white individuals (8.1 and 49.8), Hispanics (4.3 and 21.7), or Asians (3.2 and 18.9). Women were two times more likely to have sarcoidosis, with the highest prevalence for sarcoidosis noted in African American women (178.5). The data from the National Health Center for health statistics showed the highest mortality in sarcoidosis was in women4. The average death of Black patients is between 45-54 years, whereas the average death of white patients was 65-74 years5 .
Organ Involvement
The lung is almost always involved in sarcoidosis. In an ACCESS study, about half of the patients had extrapulmonary sarcoidosis with isolated organ involvement except pulmonary involvement was only 2%. Women have more extrapulmonary involvement than men. The most common organs affected by sarcoidosis other than lungs are skin, including erythema nodosum followed by liver, peripheral lymph node, eye, brain, heart, and joint6 .
Bone and Joint Involvement
About one-quarter to one-third of patients with sarcoidosis have musculoskeletal manifestation with joint, bone, and muscle involvement. A rheumatologist may encounter sarcoidosis in several situations: when patients present with primary musculoskeletal symptoms; when a patient with established sarcoidosis develops new musculoskeletal symptoms; or when other services need advice for immunosuppression.
Acute Arthritis (Löfgren Syndrome)
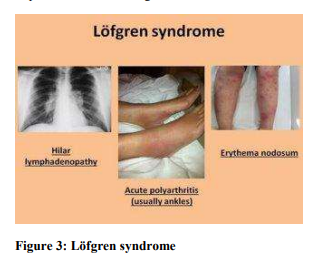
Acute arthritis is the most common musculoskeletal manifestation of sarcoidosis and can be part of Löfgren syndrome (Figure 3). This syndrome is a triad which includes symmetrical hilar adenopathy, joint pain, and erythema nodosum. There is a strong association with the HLA-DRB1 and HLA-DRB1*03. Chest radiographs reveal bilateral hilar adenopathy in 90% of patients. The most common joint involved in Löfgren syndrome are the ankles; however, other joints which can be involved are knees, wrist, elbows and metacarpophalangeal joints. True joint synovitis is rare in sarcoidosis. Patients may present with joint swelling—usually due to periarticular soft tissue swelling and tenosynovitis. Nonetheless, the prognosis of this syndrome is excellent with complete resolution usually occurring in a few months. The clinical syndrome of Löfgren syndrome is so characteristic that it is usually sufficient to reach the diagnosis of sarcoidosis. Therefore, Löfgren syndrome is one of the few scenarios where histology is not required to make the diagnosis7 .
Chronic Arthritis
It is usually present when there are other extrapulmonary manifestations, mostly the skin. The typical pattern of chronic arthritis is symmetric medium to large joint oligoarthritis. Tenosynovitis is more common than true synovitis. There is high suspicion of musculoskeletal sarcoidosis when there are symmetric inflammatory lesions of the extensor tendon compartment of the wrists. Destructive arthritis is less frequent in sarcoidosis. When there is true synovitis with large joint involvement other possibilities should be explored. Synovial tissue biopsy is warranted to demonstrate granuloma.7
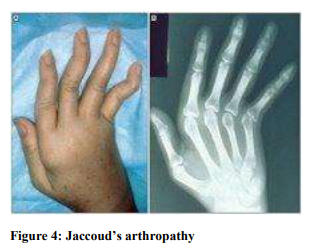
Jaccoud’s Arthropathy (Figure 4)
Jaccoud’s arthropathy is characterized by deforming (but not erosive) arthritis. It is usually present when there is extensive internal organ involvement by sarcoidosis. Biopsy shows fibrosis of tendon with presence of granuloma in the muscles and tendon sheets.7
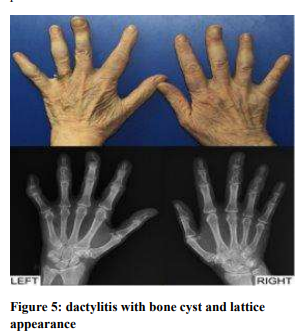
Dactylitis (Figure 5)
Dactylitis is one of the most familiar presentations of musculoskeletal involvement of sarcoidosis and causes swelling and erythema. It is almost always present with chronic systemic involvement. Dactylitis is usually symmetrical and most often affects second and third phalanges, usually the spare metacarpophalangeal joint. Biopsy shows tenosynovitis and soft tissue granuloma. Cystic bone lesion with lattice like appearance is also characteristic in radiographic findings. Joint erosion is usually not present.7
Acute and Chronic Myopathy
Although half of all sarcoid patients might have skeletal muscle involvement, only <3% of patients might be symptomatic. Chronic myopathy, however, is more common. It is insidious in onset. Patients with chronic myopathy usually present with generalized weakness, fatigue, and reduced exercise capacity. In addition to is symmetrical proximal myopathy, trunk and neck muscles can also be involved. Although muscle enzymes might be at normal level, the actual muscle biopsy shows presence of granuloma with endomysial and perivascular inflammation. In case of acute myopathy, in contrast, the onset is rapid and usually it presents in the early course of the disease— it usually causes elevated muscle enzymes and muscle biopsy shows non-caseating granulomas7.
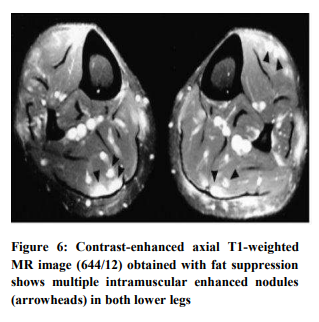
Nodular Sarcoid Myopathy
Nodular sarcoid myopathy is characterized by the presence of single or multiple nodules in the muscles. Usually there is symmetrical limb involvement. The nodules are often painful, and overtime can lead to contractures. Muscle biopsy shows the presence of lesions between muscle bundles without direct involvement of muscle fibers. MRI is a useful modality for localizing this pattern of myopathy. Nodules appear round, ovoid or fusiform, extending alongside the muscle fibers7 (Figure 6).
Sarcoid Bone Involvement
Bone involvement is usually asymptomatic and often identified following imaging for other reasons. Skin disease and soft tissue swelling are often accompanied by bone involvement. The most frequently affected bones are the proximal and middle phalanges; however, other bones can also be affected—such as skull, nasal bones, maxilla, sternum, ribs, vertebra, pelvis, tibia, and femurs. Peripheral bone involvement is more common, but more axial involvement is being diagnosed due to new imaging techniques (MRI/PET). There are three patterns of bone involvement in sarcoidosis: a “moth-eaten" appearance (which involves the cortex of the phalanges), lytic lesion— which is also called a bone cyst—which can cause cortical defect of phalangeal head; and sclerotic lesions (which are seen in the spine and look like metastatic disease). Serum calcium and alkaline phosphatase levels are typically normal despite multiple affected bones7.
Diagnosis of sarcoidosis
There is no standardized method for the diagnosis of sarcoidosis. The first evidence-based guideline for the diagnosis and detection of sarcoidosis was published in 2020 by a multidisciplinary committee of experts from American Thoracic Society. The diagnosis is based upon three major criteria: a compatible clinical presentation, finding of non–necrotizing granulomatous inflammation in one or more tissue samples, and exclusion of alternation causes granulomatous disease—including tuberculosis mycobacterium, IBD, systemic vasculitis and lymphoproliferative disorders.
The committee also decided not to do any lymph node sampling if there is strong clinical suspicion of sarcoidosis: Löfgren syndrome, Heerfordt’s syndrome, and asymptomatic bilateral hilar adenopathy. They also provided guidelines for screening of extrapulmonary sarcoidosis.
As about one fourth of patients might have ocular involvement the committee suggested a baseline eye exam. There is abnormal calcium metabolism in sarcoidosis. Sometimes the patients might have asymptomatic hypercalcemia and hypercalciuria, which can lead to nephrolithiasis and renal failure. As a result, the committee suggested baseline serum calcium level testing. They recommended checking both 25 and 1, 25 –OH vitamin D for evaluating risks and benefits of calcium and vitamin D supplement. Because hematological abnormalities like anemia and leucopenia are common, the committee has suggested doing baseline complete blood cell count to screen hematologic abnormalities8. Serum angiotensin– converting enzyme (ACE) level is widely used for diagnosis of sarcoidosis but it is not either sensitive or specific7.
Imaging modalities are helpful for diagnosis of extent of organ involvement. Lesions show increased uptake of fluorodeoxyglucose (FDG) and thus PET-CT. Cardiac MRI or Cardiac PET scan can also be used in suspected cardiac involvement. TTE has low sensitivity in diagnosis of cardiac sarcoidosis. Combination of serum biomarkers like ACE, lysozyme can be used as a prognostic tool. Imaging modalities (HRCT, PET scan) also can be used to assess the disease progression9.
Diagnosis of acute arthritis in sarcoidosis (Löfgren syndrome) is clinical. The key manifestation of chronic arthritis in sarcoidosis is be non-deforming arthritis with granulomatous synovitis, Jaccoud’s type deformity of the hands, joint swelling adjacent to sarcoid bone lesion, and dactylitis. When the patient does not have a history of sarcoidosis but has joint symptoms we diagnose the case with strong clinical suspicion, elevated biomarker (ACE level, lysozyme), and synovial biopsy (if needed) after excluding all other possible etiologies.
Treatment of Sarcoid Arthropathy
There are no society guidelines for the treatment of sarcoid arthropathy. It is mostly clinical and evidencebased. The first line treatment of acute sarcoid arthropathy is anti-inflammatory therapy with nonsteroidal anti-inflammatory drugs (NSAIDS). If the response is not adequate, we can start trial of low dose steroid10 (10 to 20mg per day) until adequate response. After that, we can then taper the dose—the goal is to decrease the dose to less than 7.5mg daily without any recurrent symptoms.
If the patients do not respond completely with the above treatment or if the prednisone dose cannot be tapered off due to frequent flare, we can add hydroxychloroquine. In patients who have some improvement of the symptoms—but continue to have arthritis symptoms after 3 months of treatment with hydroxychloroquine with low dose prednisone—we can add colchicine 0.6mg two or three times per day.
In patients who continue to have symptoms with the combination of treatment of low dose prednisone, hydroxychloroquine, and colchicine after two or three months, we can discontinue colchicine and add a more potent conventional non biologic DMARDS like methotrexate with prednisone and hydroxychloroquine. The use of methotrexate in sarcoid arthropathy is supported by limited case series and clinical experience11.
When the symptoms continue with methotrexate and low dose prednisone, we can start biologic DMARDS. TNF-alpha plays an important role in the formation of granuloma in sarcoidosis. Thus, TNF Bloker is a potential therapeutic target7. Multiple case reports and randomized trials showed the effectiveness of Infliximab, a TNF-alpha blocker, in treatment of resistant pulmonary and extrapulmonary sarcoidosis12.
There are no randomized controlled trials for the treatment of sarcoid myopathy. Corticosteroid is generally effective in treating acute and nodular muscular sarcoidosis; however these are not as efficacious for the treatment of chronic sarcoid myopathy.7 We generally continue the effective regimen for about one year before initiating the gradual tapering of medication over 6-12 months, depending upon the clinical response of the patients.
Prognosis
The outcome of both acute and chronic sarcoidosis is favorable. Sometimes acute sarcoidosis is self-limiting and resolves without any permanent deformity. Occasionally, however, acute sarcoidosis may recur and can develop chronic sarcoidosis of the lung13. Though the prevalence of chronic sarcoid arthritis is not common, it can cause joint deformities.
1. Ungprasert P, Ryu JH, Matteson EL. Clinical Manifestations, Diagnosis, and Treatment of Sarcoidosis. Mayo Clin Proc Innov Qual Outcomes. 2019; 3 (3):358-375. Published 2019 Aug 2. doi:10.1016/j.mayocpiqo.2019.04.006
2. DANBOLT N. The historical aspects of sarcoidosis. Postgrad Med J. 1958 May; 34 (391):245-7 passim. doi: 10.1136/pgmj.34.391.245. PMID: 13553999; PMCID: PMC2501582.
3. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New England Journal of Medicine. 2007; 357(21):2153-2165. Doi: 10.1056/nejmra071714
4. Baughman RP, Field S, Costabel U, Crystal RG, Culver DA, Drent M, Judson MA, Wolff G. Sarcoidosis in America. Analysis Based on Health Care Use. Ann Am Thorac Soc. 2016 Aug; 13 (8):1244-52. doi: 10.1513/AnnalsATS.201511- 760OC. PMID: 27509154.
5. Swigris JJ, Olson AL, Huie TJ, Fernandez-Perez ER, Solomon J, Sprunger D, Brown KK. Sarcoidosisrelated mortality in the United States from 1988 to 2007. Am J Respir Crit Care Med. 2011 Jun 1; 183 (11):1524-30. doi: 10.1164/rccm.201010-1679OC. Epub 2011 Feb 17. PMID: 21330454; PMCID: PMC3137141.
6. Aksoy E, Tunçay E, Ocakl B. Extrapulmonary Sarcoidosis with Multiple-Organ Involvement. Southern Clinics of Istanbul Eurasia. Published online 2018.
7. Bechman K, Christidis D, Walsh S, Birring SS, Galloway J. A review of the musculoskeletal manifestations of sarcoidosis. Rheumatology (Oxford). 2018 May 1; 57(5):777-783. doi: 10.1093/rheumatology/kex317. PMID: 28968840.
8. Singha A, Liao SY, Herman DD, Crouser ED, Maier LA, Baughman RP et al. Summary for Clinicians: Clinical Practice Guideline for the Diagnosis and Detection of Sarcoidosis. Annals of the American Thoracic Society 2020; 17:1510-1515. doi: 10.1164/rccm.202002-0251ST .
9. Kraaijvanger R, Janssen Bonás M, Vorselaars ADM, Veltkamp M. Biomarkers in the Diagnosis and Prognosis of Sarcoidosis: Current Use and Future Prospects. Front Immunol. 2020 Jul 14; 11:1443. doi: 10.3389/fimmu.2020.01443. PMID: 32760396; PMCID: PMC7372102.
10. Sweiss NJ, Patterson K, Sawaqed R et al. Rheumatologic manifestations of sarcoidosis. Semin Respir Crit Care Med 2010; 31:46373
11. Kaye O, Palazzo E, Grossin M, Bourgeois P, Kahn MF, Malaise MG. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol. 1995 Jul; 34 (7): 642-4. doi: 10.1093/rheumatology/34.7.642. PMID: 7670783.
12. Judson MA, Baughman RP, Costabel U, Flavin S, Lo KH, Kavuru MS, Drent M; Centocor T48 Sarcoidosis Investigators. Efficacy of infliximab in extrapulmonary sarcoidosis: results from a randomised trial. Eur Respir J. 2008 Jun; 31(6):1189- 96. doi: 10.1183/09031936.00051907. Epub 2008 Feb 6. PMID: 18256069.
13. Gran JT, Bøhmer E. Acute sarcoid arthritis: a favourable outcome? A retrospective survey of 49 patients with review of the literature. Scand J Rheumatol. 1996; 25(2):70-3. doi: 10.3109/03009749609069210. PMID: 8614769.